In this study, we demonstrated that inhibition of PI3Ks promoted nocodazole-induced mitotic cell death and reduced mitotic slippage. This finding suggests that using PI3k inhibitors in combination with anti-mitotic drugs may improve cancer treatment outcomes. In summary, the current study demonstrated that the inhibition of PI3K pathway induced mitotic arrest and mitotic cell death and promoted nocodazole-induced mitotic cell death while reducing the occurrence of mitotic slippage. These results suggest a novel role for the PI3K pathway in regulating cell cycle progression during mitosis and preventing mitotic cell death, and provide justification for the use of PI3K inhibitors in combination with anti-mitotic drugs to combat cancer. Isoprenoids constitute one of the largest groups of natural product compounds. They are structurally diverse and include cannabinoids, essential oils, sterols, the prenyl groups of chlorophyll and RNA among others. Isoprenoids are involved in respiration, hormone-based signalling, the post-translational processes that control lipid biosynthesis, meiosis, apoptosis, glycoprotein biosynthesis, and protein degradation. Furthermore, they represent important structural components of cell membranes. All isoprenoids are synthesised from two simple precursors, isopentenyl pyrophosphate and dimethylallyl pyrophosphate. The precursors are provided by two distinct biosynthetic pathways, which are distributed in an organism specific manner. In mammals, the plant cytosol, certain bacteria and trypanosomatids, these compounds are products of the mevalonate pathway. In most eubacteria, algae, chloroplasts, cyanobacteria and apicomplexan parasites the deoxy-xylulose phosphate pathway generates IPP and DMAPP. This biosynthetic route to isoprenoid precursors is an essential aspect of metabolism and the DOXP pathway is a genetically validated target for broad-spectrum antimicrobial drugs against malaria, tuberculosis, and a range of sexually transmitted conditions. The absence of this pathway in humans makes it a particular attractive target for antimicrobial drug discovery. Chemical validation is provided by the anti-malarial compound fosmidomycin, which inhibits 1-deoxy-D-xylulose 5-phosphate reductoisomerase. Two types of IspE inhibitors are known. The majority of IspE inhibitors mimic either the cytidine or phosphate-sugar moiety of the substrate CDP-ME. Crystal structures of AaIspE in complex with cytidine analogues containing a benzimidazole moiety attached to the ribose have been determined suggesting that interactions in the cytidine Y-27632 dihydrochloride ROCK inhibitor pocket are key for binding affinity. Considering the size of these molecules they are rather weak ligands for BU 4061T EcIspE with affinities in the double-digit micromolar range. In contrast, the smaller cytosine analogues bind more tightly to EcIspE with some compounds of this series displaying IC50 values in the low micromolar range. Very recently, non-substrate like EcIspE inhibitors have been reported. The best characterized compounds also have IC50 values in the low micromolar range. They were proposed to bind into the substrate binding site forming stacking interactions with Tyr25 and Phe185; however, the derived structure-activity relationships were not always consistent with this binding mode as large changes to the presumably pistacking moieties did not lead to large changes in affinity. Motivated by the potential of IspE as a target for broadspectrum antimicrobial drugs we sought to discover non-substrate like IspE inhibitors that can 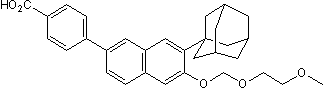 serve as starting points for the development of new antimicrobials. There are several methods for hit discovery.
serve as starting points for the development of new antimicrobials. There are several methods for hit discovery.
Elucidation of the signaling pathway during prolonged mitotic arrest is important to improve the effects of anti-mitotic drugs
Leave a reply
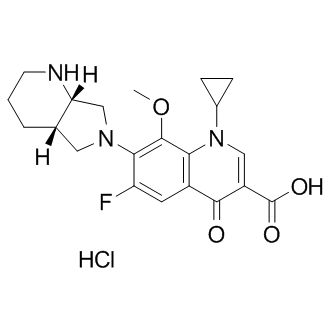 a template for structurebased design of inhibitors with broad-spectrum antimicrobial activity. However, since we had been able to reproducibly crystallize and gain most crystallographic information with
a template for structurebased design of inhibitors with broad-spectrum antimicrobial activity. However, since we had been able to reproducibly crystallize and gain most crystallographic information with  rings. The most improved inhibitors identified in the 7-carbon series have their meta- and para-substituted methoxyl and hydroxyl groups reversed from that of curcumin, as with compound 1, or methoxyl groups placed in both positions, as with compound 2. The simple substitution of the para-hydroxy group on curcumin with a methoxy substitution improved inhibitor function by 6-7-fold over that measured for curcumin, making compound 2 our most potent lead analog for anti-Ab aggregation activity. Additional challenges lie ahead to improve the bioactivity of our curcumin-derived analog in order to increase the therapeutic dose to the CNS. Questions in regard to bioavailability have plagued the use of curcumin as a potential therapeutic for a number of years. Clinical trials have shown that the inherent bioavailability of orally administered curcumin is relatively low when factoring in intestinal absorption, liver metabolism and BBB penetrance. However, in spite of these difficulties, dietary supplementation of curcumin administered to aged APPtransgenic mice significantly lowered Ab deposition in the CNS. These findings clearly show that curcumin is able to enter the circulation and cross the BBB in sufficient quantities to reduce amyloid burden. To improve upon this property, we anticipate that the methoxy substitution on our lead compound 2 will decrease polarity and increase lipid membrane solubility thereby improving passive diffusion across the blood brain barrier and access to the CNS. Similar observations have been made for other inhibitors of Ab aggregation such as Chrysamine G. In this study, the more lipophilic compound Chrysamine G was compared with Congo Red and found to readily cross the BBB in normal mice, achieving a brain:blood ratio of greater than 10:1. Moreover, metabolic inactivation poses other challenges to maintaining bioactivity. In this respect, the hydroxyl groups on curcumin are modified by enzymes found in the liver, kidney and intestinal mucosa to form curcumin glucuronides and curcumin sulfates. The methoxy substitution for these hydroxyl groups on our lead compound 2 should prevent these glucuronide and sulfate additions and contribute to sustained bioactivity.
rings. The most improved inhibitors identified in the 7-carbon series have their meta- and para-substituted methoxyl and hydroxyl groups reversed from that of curcumin, as with compound 1, or methoxyl groups placed in both positions, as with compound 2. The simple substitution of the para-hydroxy group on curcumin with a methoxy substitution improved inhibitor function by 6-7-fold over that measured for curcumin, making compound 2 our most potent lead analog for anti-Ab aggregation activity. Additional challenges lie ahead to improve the bioactivity of our curcumin-derived analog in order to increase the therapeutic dose to the CNS. Questions in regard to bioavailability have plagued the use of curcumin as a potential therapeutic for a number of years. Clinical trials have shown that the inherent bioavailability of orally administered curcumin is relatively low when factoring in intestinal absorption, liver metabolism and BBB penetrance. However, in spite of these difficulties, dietary supplementation of curcumin administered to aged APPtransgenic mice significantly lowered Ab deposition in the CNS. These findings clearly show that curcumin is able to enter the circulation and cross the BBB in sufficient quantities to reduce amyloid burden. To improve upon this property, we anticipate that the methoxy substitution on our lead compound 2 will decrease polarity and increase lipid membrane solubility thereby improving passive diffusion across the blood brain barrier and access to the CNS. Similar observations have been made for other inhibitors of Ab aggregation such as Chrysamine G. In this study, the more lipophilic compound Chrysamine G was compared with Congo Red and found to readily cross the BBB in normal mice, achieving a brain:blood ratio of greater than 10:1. Moreover, metabolic inactivation poses other challenges to maintaining bioactivity. In this respect, the hydroxyl groups on curcumin are modified by enzymes found in the liver, kidney and intestinal mucosa to form curcumin glucuronides and curcumin sulfates. The methoxy substitution for these hydroxyl groups on our lead compound 2 should prevent these glucuronide and sulfate additions and contribute to sustained bioactivity. small scaffolds led to the
small scaffolds led to the