Although confirmation of the efficacy of this pharmacological approach awaits completion of large clinical trials, the adjuvant use of these compounds is common in patients that do not meet targeted reductions of lipoproteins while taking statins. Given the high prevalence of lipid metabolism disorders it is desirable to identify lead compounds that can be developed into new drugs that inhibit lipid absorption via novel mechanisms. Here we report the utility of using the zebrafish for this purpose. Because of their small size, optical transparency zebrafish larvae are well suited for chemical library screens using fluorescent, histochemical or morphological assays. Indeed, a great advantage of chemical screens in zebrafish is the ability to rapidly assess compound efficacy and toxicity in vivo. Given the high degree of conservation of lipid metabolism in teleost fish and mammals, it is likely that compounds identified in a zebrafish screen will act through comparable mechanisms in mammals. Here we report the results of a pilot screen of a non-biased chemical library through which we identified 7 novel compounds that inhibited the absorption of fluorescent lipid analogues. We show that compounds identified in the primary screening assay can be rapidly prioritized for testing in mammals using a variety of simple, yet Everolimus highly informative in vivo secondary assays. The secondary assays also Torin 1 company provided insights into the compounds�� mechanism of action, which could be 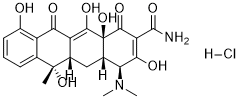 distinguished from the effects of orlistat and ezetimibe in zebrafish larvae. Surprisingly, we found that ezetimibe inhibited absorption of not only cholesterol analog, but also long chain fatty acid and phopholipid analogs. Together, these findings demonstrate the feasibility of conducting screens for compounds that interfere with complex physiological processes using the zebrafish. The screening assays used for this study were derived from previous work using fluorescent lipid reporters in zebrafish larvae. Following their ingestion, the fluorescent metabolites of these reporters are first detected in the gallbladder of live larvae and later the intestinal lumen following gallbladder contraction. The compounds are used at low concentrations and they are rapidly absorbed from the intestinal lumen, thus their fluorescence emission is not detected in the intestinal lumen immediately after ingestion or when absorption in inhibited. Fluorescence emission from one of the analogues, the phospholipid PED-6, is quenched prior to metabolism by luminal phospholipase. Thin layer chromatographic analyses of bile from adult fish, or total body lipids of 5 dpf larvae, showed that PED-6, which is labeled with a BODIPY labeled short chain fatty acid at the sn-2 position, is metabolized to cholesterol esters, phospholipids and possibly, triglyceride. Free PED6 was not detected in either assay. For the primary screen, 5 day post-fertilization larvae were arrayed in 96 well plates and soaked overnight in test compounds. The following morning larvae were soaked in PED-6 for 6 hours after which a qualitative visual assessment of gallbladder fluorescence was made using an inverted compound microscope. Reduced gallbladder fluorescence, the endpoint we use to identify active compounds in the primary screen, could not differentiate compounds that inhibited lipid absorption from those that interfered with swallowing, phospholipase activity or hepatic metabolism and biliary secretion. As described below, secondary assays were devised to distinguish these mechanistic possibilities.
distinguished from the effects of orlistat and ezetimibe in zebrafish larvae. Surprisingly, we found that ezetimibe inhibited absorption of not only cholesterol analog, but also long chain fatty acid and phopholipid analogs. Together, these findings demonstrate the feasibility of conducting screens for compounds that interfere with complex physiological processes using the zebrafish. The screening assays used for this study were derived from previous work using fluorescent lipid reporters in zebrafish larvae. Following their ingestion, the fluorescent metabolites of these reporters are first detected in the gallbladder of live larvae and later the intestinal lumen following gallbladder contraction. The compounds are used at low concentrations and they are rapidly absorbed from the intestinal lumen, thus their fluorescence emission is not detected in the intestinal lumen immediately after ingestion or when absorption in inhibited. Fluorescence emission from one of the analogues, the phospholipid PED-6, is quenched prior to metabolism by luminal phospholipase. Thin layer chromatographic analyses of bile from adult fish, or total body lipids of 5 dpf larvae, showed that PED-6, which is labeled with a BODIPY labeled short chain fatty acid at the sn-2 position, is metabolized to cholesterol esters, phospholipids and possibly, triglyceride. Free PED6 was not detected in either assay. For the primary screen, 5 day post-fertilization larvae were arrayed in 96 well plates and soaked overnight in test compounds. The following morning larvae were soaked in PED-6 for 6 hours after which a qualitative visual assessment of gallbladder fluorescence was made using an inverted compound microscope. Reduced gallbladder fluorescence, the endpoint we use to identify active compounds in the primary screen, could not differentiate compounds that inhibited lipid absorption from those that interfered with swallowing, phospholipase activity or hepatic metabolism and biliary secretion. As described below, secondary assays were devised to distinguish these mechanistic possibilities.
Therefore may be used as an adjuvant or alternative to HMG co-reductase inhibitors for the primary
Leave a reply
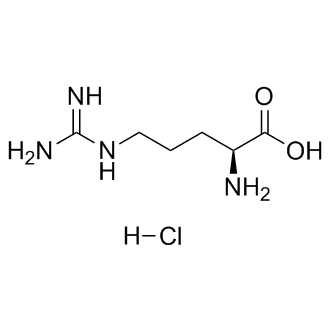 atom. Deviating slightly from the binding mode of CKI-7 to APH-Ia, the contact between the Nb of the aminoethyl and carbonyl of Leu88 located in the linker of the enzyme is achieved via a water molecule, compared to a direct interaction observed in APH-Ia. Hemostasis is one of the most important processes in organisms, and disorders in this system cause deaths under a variety of pathologies. The activation of blood coagulation can be caused by trauma, sepsis, inflammation, obstetric practice and in the course of
atom. Deviating slightly from the binding mode of CKI-7 to APH-Ia, the contact between the Nb of the aminoethyl and carbonyl of Leu88 located in the linker of the enzyme is achieved via a water molecule, compared to a direct interaction observed in APH-Ia. Hemostasis is one of the most important processes in organisms, and disorders in this system cause deaths under a variety of pathologies. The activation of blood coagulation can be caused by trauma, sepsis, inflammation, obstetric practice and in the course of 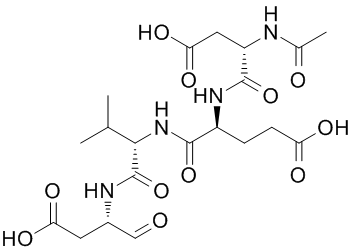 antibiotics in pathogens has been exacerbated by the lack of novel antibacterials being introduced to the market. An alternative and parallel approach in supporting the mitigation of the antibiotic resistance problem is to develop adjuvants that could interfere with the mechanism of resistance and hence restore the action of antibiotics. Such a strategy has been effectively employed to combat resistance to b-lactams due to b-lactamase activity. For aminoglycosides, a group of antibiotics used to treat serious nosocomial infections, the main mechanism of resistance is via the enzymatic inactivation of the drug by acetyltransferases, nucleotidyltransferases, or phosphotransferases. This implies that inhibitors of these enzymes could be exploited for the development of drug-adjuvant therapy. Among the three types of aminoglycoside-modifying enzymes, aminoglycoside phosphotransferases or kinases yield the highest levels of resistance thereby providing a rationale for focusing inhibitor development for these specific resistance factors. The investigation of APH inhibitors that target the ATP-binding pocket was facilitated by the structural similarities between the aminoglycoside resistance enzyme APH-IIIa and serine/threonine and tyrosine eukaryotic protein kinases, especially in the Nterminal lobe. It was subsequently shown that APH-IIIa can be inhibited by protein kinase inhibitors of the isoquinolinesulfonamide family and they are competitive with ATPbinding. However, APH-IIIa remains the most extensively studied due to its broad substrate spectrum.
antibiotics in pathogens has been exacerbated by the lack of novel antibacterials being introduced to the market. An alternative and parallel approach in supporting the mitigation of the antibiotic resistance problem is to develop adjuvants that could interfere with the mechanism of resistance and hence restore the action of antibiotics. Such a strategy has been effectively employed to combat resistance to b-lactams due to b-lactamase activity. For aminoglycosides, a group of antibiotics used to treat serious nosocomial infections, the main mechanism of resistance is via the enzymatic inactivation of the drug by acetyltransferases, nucleotidyltransferases, or phosphotransferases. This implies that inhibitors of these enzymes could be exploited for the development of drug-adjuvant therapy. Among the three types of aminoglycoside-modifying enzymes, aminoglycoside phosphotransferases or kinases yield the highest levels of resistance thereby providing a rationale for focusing inhibitor development for these specific resistance factors. The investigation of APH inhibitors that target the ATP-binding pocket was facilitated by the structural similarities between the aminoglycoside resistance enzyme APH-IIIa and serine/threonine and tyrosine eukaryotic protein kinases, especially in the Nterminal lobe. It was subsequently shown that APH-IIIa can be inhibited by protein kinase inhibitors of the isoquinolinesulfonamide family and they are competitive with ATPbinding. However, APH-IIIa remains the most extensively studied due to its broad substrate spectrum.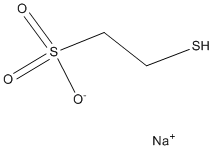 and the availability of potential targets. The affinity we measured between IkB and TCL1A was about 1,000 fold weaker than the one previously reported between IkB and NF-kB.
and the availability of potential targets. The affinity we measured between IkB and TCL1A was about 1,000 fold weaker than the one previously reported between IkB and NF-kB.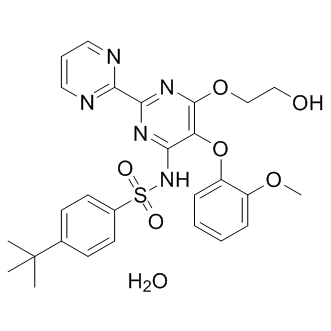 combined effects of BOR/PSI with IM on CML in vivo and in vitro. Intriguingly, the results showed that the combinatory regimens yielded enhanced therapeutic efficacies in CML murine models, potentiated effects on CML cells, and triggered
combined effects of BOR/PSI with IM on CML in vivo and in vitro. Intriguingly, the results showed that the combinatory regimens yielded enhanced therapeutic efficacies in CML murine models, potentiated effects on CML cells, and triggered