According to unpublished data quoted by Inutsuka et al., SOB has been shown to be protective against DOX cardiac and renal toxicity in rats. Additionally, SOB exhibited synergy with DOX in colon-26 tumour-bearing mice as measured by prolonged survival of the animals. In the same study, the authors observed an increased G2/M population after incubating colon-26 cells with a combination of SOB and DOX for 24 h compared to DOX-treated samples. The G2/M arrest was also reported in a human mammary tumour cell line exposed to 4 mM SOB for 20 h. Given the gradual increase in ANT sensitivity as cells transition from relatively resistant G1 phase to G2/M phase, which was observed in exponentially growing Molt-4 cells, the accumulation of G2/M cells before ANT treatment could be beneficial as the relatively resistant population is depleted in favour of the more sensitive population. In our study, SOB alone did not induce significant G2/M phase arrest of HL-60 cells after treatment for 72 h. However, SOB significantly reduced the G1 and S phase populations, which could be favourable for increasing HL-60 cell sensitivity to DOX. The combination of SOB with DOX caused even more pronounced depletion of G1 and S phase cells together with an increase in polyploid cells and cell debris. Indeed, the combination of DOX or DAU with SOB in our study provoked unequivocally synergistic antiproliferative effects in HL-60 cells at all assessed concentrations and schedule. Conversely, SOB pre-incubation with NVCMs led to significant protection from DAU- and DOXinduced toxicity as assessed by LDH release, mitochondrial membrane potential measurements and caspase activity measurements. MER is a non-sedative barbiturate derivative that is structurally unrelated to bisdioxopiperazines. Its principal effect on replicating cells is the inhibition of chromosome condensation, activation of c-Jun and JNKs and induction of G2/M cell cycle blockade. In this study, although MER significantly protected NVCMs against both DAU- and DOX-induced LDH release, it also induced some toxicity in NVCMs. MER insignificantly reduced the DAU-induced activation of caspases and also only partially protected cardiomyocytes from DYm loss. However, of the three catalytic Vorinostat 149647-78-9 inhibitors examined in this study, MER showed the highest potential to act synergistically with ANTs in HL-60 cells, particularly at higher concentrations for which there was no tendency towards increased CI values. Additionally, with 3- or 6-h pre-incubations, the drug combinations remained consistently synergistic. The effects of MER on the cell cycle in HL-60 cells differed considerably from the bisdioxopiperazines. MER alone caused dramatic G2/M arrest, which was even more pronounced than that induced by DOX. In addition, the sub G1 and polynuclear populations increased. However, unlike DEX or SOB, the MER + DOX combination did not induce more pronounced cell cycle changes. As DEX and SOB are the members of the bis-dioxopiperazine family, they can be metabolized to 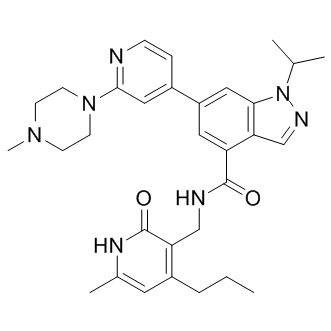 Fe-chelating compounds. Whereas DEX is well known to undergo metabolization to ADR925, SOB must be LY2109761 side effects probably first metabolized to ICRF-154 and then to the open-ring product, thus requiring two-step metabolism to metal chelator. To examine this issue, the Fe intracellular chelating properties of DEX, SOB and also MER were examined in H9c2 rat embryonic cardiomyoblast-derived cell line using the measurements of calcein-AM fluorescence intensity to compare the cardioprotection with their ability to chelate Fe. The experimental strong lipophilic Fe chelator SIH was repeatedly documented to chelate free Fe both in solution as well as in cells, and to displace Fe from its complex with calcein resulting in dequenching of its fluorescence and therefore it has been used as the reference chelator in our study. Indeed, fast increase in calcein fluorescence could be observed upon addition of 100 mM SIH to the cells with intracellular-trapped calcein-Fe complex and also DEX is able to significantly chelate Fe in comparison with control, although rather slowly and with only,30% efficiency as compared to SIH. The,2.5 h lag-time before the start of fluorescence increase was probably caused by the need of DEX hydrolysis to the chelating metabolite ADR-925. Similar result was observed by Hasinoff et al., who found that unlike ADR-925, which displaces Fe from the cell-trapped calcein-Fe complex in a pattern similar to SIH, DEX chelated Fe only partially and with substantial lag-time.
Fe-chelating compounds. Whereas DEX is well known to undergo metabolization to ADR925, SOB must be LY2109761 side effects probably first metabolized to ICRF-154 and then to the open-ring product, thus requiring two-step metabolism to metal chelator. To examine this issue, the Fe intracellular chelating properties of DEX, SOB and also MER were examined in H9c2 rat embryonic cardiomyoblast-derived cell line using the measurements of calcein-AM fluorescence intensity to compare the cardioprotection with their ability to chelate Fe. The experimental strong lipophilic Fe chelator SIH was repeatedly documented to chelate free Fe both in solution as well as in cells, and to displace Fe from its complex with calcein resulting in dequenching of its fluorescence and therefore it has been used as the reference chelator in our study. Indeed, fast increase in calcein fluorescence could be observed upon addition of 100 mM SIH to the cells with intracellular-trapped calcein-Fe complex and also DEX is able to significantly chelate Fe in comparison with control, although rather slowly and with only,30% efficiency as compared to SIH. The,2.5 h lag-time before the start of fluorescence increase was probably caused by the need of DEX hydrolysis to the chelating metabolite ADR-925. Similar result was observed by Hasinoff et al., who found that unlike ADR-925, which displaces Fe from the cell-trapped calcein-Fe complex in a pattern similar to SIH, DEX chelated Fe only partially and with substantial lag-time.
Enhanced the activation of caspases induced by DOX in particular malignant lymphoma and adult T-cell leukaemia
Leave a reply
 has better binding towards the mutation T315A than T315I. The free energy of BCR-ABLT315I
has better binding towards the mutation T315A than T315I. The free energy of BCR-ABLT315I  subtilis EzrAhave separable, yet overlapping roles in mediating EzrA’s interaction with FtsZ and potentially other cellular
subtilis EzrAhave separable, yet overlapping roles in mediating EzrA’s interaction with FtsZ and potentially other cellular  already reached the stage of
already reached the stage of  of the positive and the negative training datasets. This was observed with all the types of features. For an experimenter, a judicious approach would be minimizing the number of CDKIs to be characterized by increasing the threshold to higher SVM score, in order to get only the topmost candidates for further work. Supplementing these with other complementary evidence like domain
of the positive and the negative training datasets. This was observed with all the types of features. For an experimenter, a judicious approach would be minimizing the number of CDKIs to be characterized by increasing the threshold to higher SVM score, in order to get only the topmost candidates for further work. Supplementing these with other complementary evidence like domain