However, at term, placental and fetal weights are strongly correlated among the normosomic newborns from N-STZ or control dams. In contrast, this linear relationship does not exist in macrosomic pups, which is indicative of the disharmonious development of the fetus and its placenta. Furthermore, histological analysis revealed the overall higher size and enlarged labyrinthine layer in the macrosomic placentas compared with normosomic placentas, whereas the spongiotro phoblast size was not significantly altered. Reduced labyrinth size and impaired spongiotrophoblast differentiation have been recently reported in diabetic mouse placentas. The authors suggested that these alterations which appear at temporally distinct phases during gestation could contribute to reduced fetal growth described in this animal model of diabetic pregnancy. Our data do not unequivocally Salvianolic-acid-B demonstrate that the rise in labyrinthine zone is responsible for fetal overgrowth, because the possibility that changes occur at early stages of gestation has not been evaluated in the current study. The molecular mechanisms underlying fetal overgrowth in pregnancies complicated with mild hyperglycemia had not yet been investigated. This is the first study that explores the expression of genes known to be 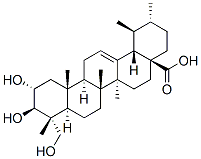 involved in the placental development and fetal growth in a model of MGH. We report that in the N-STZ pregnancies with a higher risk to have macrosomic newborns, the expression of IGF2, IGFR2 and IRS1 genes in the placentas was down regulated compared to controls. While the placentas from macrosomic pups have a reduced IRS1 gene expression, those of normotrophes were associated with a down regulation of IGF2 and IGFR2 genes. Insulin/IGFs system constitutes a major determinant for the growth of the placenta and has been shown to play a significant role in the placental adaptations in response to the nutritional challenges in several rodent species. In accordance, we show that the placental weight was higher in macrosomic newborns but also in normotrophes ones. We also found that the expression of GLUT1 gene is decreased in the N-STZ placentas of macrosomic and normosomic pups. The effects of maternal diabetes on the placental expression of Chloroquine Phosphate glucose transporters were often contradictory, and not linked to fetal growth. The reasons for these discrepancies may be explained by the temporal expression of glucose transporters isoforms during the gestation facing to the gestational period at which the analyses were conducted. For example, high fat diet in mice before and during the pregnancy increases transplacental transport of glucose and causes up regulation of protein expression of GLUT1 in the microvillous plasma membranes from placentas at embryonic day 18.5. Furthermore, these changes were associated with fetal overgrowth at this stage of development suggesting that they could be a cause of altered fetal growth. However, the high fat diet induced maternal obesity that was correlated with increased circulating leptin and decreased serum adiponectin concentrations, but normal glucose tolerance. Thereby, maternal metabolic profile was strongly different from that of the N-STZ mothers and could expose the feto-placental unit to a hormonal environment distinct because the leptin and adiponectin have been reported to regulate the transfer of nutriments through the placenta. Taken together, our data demonstrate that moderate hyperglycemia and/or glucose intolerance during pregnancy modulate the expression of several genes involved in the placental development and glucose transport without necessarily having an impact on weight at birth.
involved in the placental development and fetal growth in a model of MGH. We report that in the N-STZ pregnancies with a higher risk to have macrosomic newborns, the expression of IGF2, IGFR2 and IRS1 genes in the placentas was down regulated compared to controls. While the placentas from macrosomic pups have a reduced IRS1 gene expression, those of normotrophes were associated with a down regulation of IGF2 and IGFR2 genes. Insulin/IGFs system constitutes a major determinant for the growth of the placenta and has been shown to play a significant role in the placental adaptations in response to the nutritional challenges in several rodent species. In accordance, we show that the placental weight was higher in macrosomic newborns but also in normotrophes ones. We also found that the expression of GLUT1 gene is decreased in the N-STZ placentas of macrosomic and normosomic pups. The effects of maternal diabetes on the placental expression of Chloroquine Phosphate glucose transporters were often contradictory, and not linked to fetal growth. The reasons for these discrepancies may be explained by the temporal expression of glucose transporters isoforms during the gestation facing to the gestational period at which the analyses were conducted. For example, high fat diet in mice before and during the pregnancy increases transplacental transport of glucose and causes up regulation of protein expression of GLUT1 in the microvillous plasma membranes from placentas at embryonic day 18.5. Furthermore, these changes were associated with fetal overgrowth at this stage of development suggesting that they could be a cause of altered fetal growth. However, the high fat diet induced maternal obesity that was correlated with increased circulating leptin and decreased serum adiponectin concentrations, but normal glucose tolerance. Thereby, maternal metabolic profile was strongly different from that of the N-STZ mothers and could expose the feto-placental unit to a hormonal environment distinct because the leptin and adiponectin have been reported to regulate the transfer of nutriments through the placenta. Taken together, our data demonstrate that moderate hyperglycemia and/or glucose intolerance during pregnancy modulate the expression of several genes involved in the placental development and glucose transport without necessarily having an impact on weight at birth.
Placental index and placental efficiency were altered in macrosomic newborns but also in normosomic newborns
Leave a reply
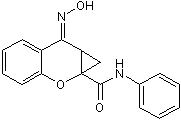 severing and rapid filament disintegration could account for additional filament deconstruction. To simulate PTEN activity, we locally disable Rac activation. When we add the PTEN activity to a fixed region, an actin wave cannot propagate
severing and rapid filament disintegration could account for additional filament deconstruction. To simulate PTEN activity, we locally disable Rac activation. When we add the PTEN activity to a fixed region, an actin wave cannot propagate 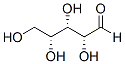 GTPases activation state by CNF1 in the two brain areas, CNF1 decreasing Rho proteins’activation in the hippocampus while activating them in the frontal cortex. This is probably due to the fact that CNF1 most certainly stimulates the activation/degradation process of Rho GTPases in both areas, but with a different outcome depending on initial activation status of Rho proteins. It is also relevant that CNF1 increases ATP availability in both hippocampus and cortex of apoE4 mice, although at different extent. How RhoA and Rac1 signaling can increase ATP is still uncharted and under investigation by our group, but we can hypothesize that the increase in ATP content observed in both brain regions could probably be linked to the CNF1-induced activation/degradation process. Furthermore, we have previously reported that CNF1 influences the mitochondrial homeostasis, induces a remarkable modification in the mitochondrial network architecture, with the appearance of elongated and interconnected mitochondria, and promotes an increment of proteins such as creatine and phosphocreatine, which are involved in ATP regeneration in the brain of pathological murine models. All these effects persist for long periods of time in mouse brains, suggesting the persistence of the CNF1 molecular effects
GTPases activation state by CNF1 in the two brain areas, CNF1 decreasing Rho proteins’activation in the hippocampus while activating them in the frontal cortex. This is probably due to the fact that CNF1 most certainly stimulates the activation/degradation process of Rho GTPases in both areas, but with a different outcome depending on initial activation status of Rho proteins. It is also relevant that CNF1 increases ATP availability in both hippocampus and cortex of apoE4 mice, although at different extent. How RhoA and Rac1 signaling can increase ATP is still uncharted and under investigation by our group, but we can hypothesize that the increase in ATP content observed in both brain regions could probably be linked to the CNF1-induced activation/degradation process. Furthermore, we have previously reported that CNF1 influences the mitochondrial homeostasis, induces a remarkable modification in the mitochondrial network architecture, with the appearance of elongated and interconnected mitochondria, and promotes an increment of proteins such as creatine and phosphocreatine, which are involved in ATP regeneration in the brain of pathological murine models. All these effects persist for long periods of time in mouse brains, suggesting the persistence of the CNF1 molecular effects 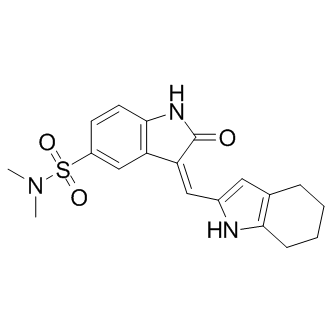 proteins of an important gut pathogen, C. jejuni. The current research emphasizes the need for future investigations in two distinct areas. First, more insight into the newly identified proteins of C. jejuni might foster the understanding of this enigmatic pathogen and help to illuminate its pathogenicity while providing suitable means for rapid detection and combating its spreading. Second, transferring this method to other bacterial pathogens will help to discover other immunodominant proteins, potentially leading to a broad spectrum of clinical applications and serological assays. Additionally, it might be preferable to simplify the identification of conformational epitopes as this could greatly enhance the retrieval rate of suitable antigenic sites.
proteins of an important gut pathogen, C. jejuni. The current research emphasizes the need for future investigations in two distinct areas. First, more insight into the newly identified proteins of C. jejuni might foster the understanding of this enigmatic pathogen and help to illuminate its pathogenicity while providing suitable means for rapid detection and combating its spreading. Second, transferring this method to other bacterial pathogens will help to discover other immunodominant proteins, potentially leading to a broad spectrum of clinical applications and serological assays. Additionally, it might be preferable to simplify the identification of conformational epitopes as this could greatly enhance the retrieval rate of suitable antigenic sites. total protein level, a defect that cannot be observed using pure analytical methodology since it cannot differentiate between intracellular and released cytokines. Misregulated cytokine release may cause chronically stressed and activated microglia, and contribute to perpetuating the inflammation and neurodegeneration seen in PD. Microglia release of critical neurotropic factors, such as NGF and BDNF and the general impact on vesicle secretion machinery by elevated levels of a-syn may be as important a contributor to disease as the alterations in inflammation. Previous studies have
total protein level, a defect that cannot be observed using pure analytical methodology since it cannot differentiate between intracellular and released cytokines. Misregulated cytokine release may cause chronically stressed and activated microglia, and contribute to perpetuating the inflammation and neurodegeneration seen in PD. Microglia release of critical neurotropic factors, such as NGF and BDNF and the general impact on vesicle secretion machinery by elevated levels of a-syn may be as important a contributor to disease as the alterations in inflammation. Previous studies have